Related Concept Videos
 01:25
01:25Nucleic Acid Structure
The pentose sugar in DNA is deoxyribose, while in RNA the pentose sugar is ribose. The difference between the sugars is the presence of the hydroxyl group on the ribose's second carbon and a hydrogen on the deoxyribose's second carbon. The phosphate residue attaches to the hydroxyl group of the 5′ carbon of one sugar and the hydroxyl group of the 3′ carbon of the sugar of the next nucleotide, which forms a 5′ to 3′ phosphodiester linkage.
DNA Structure
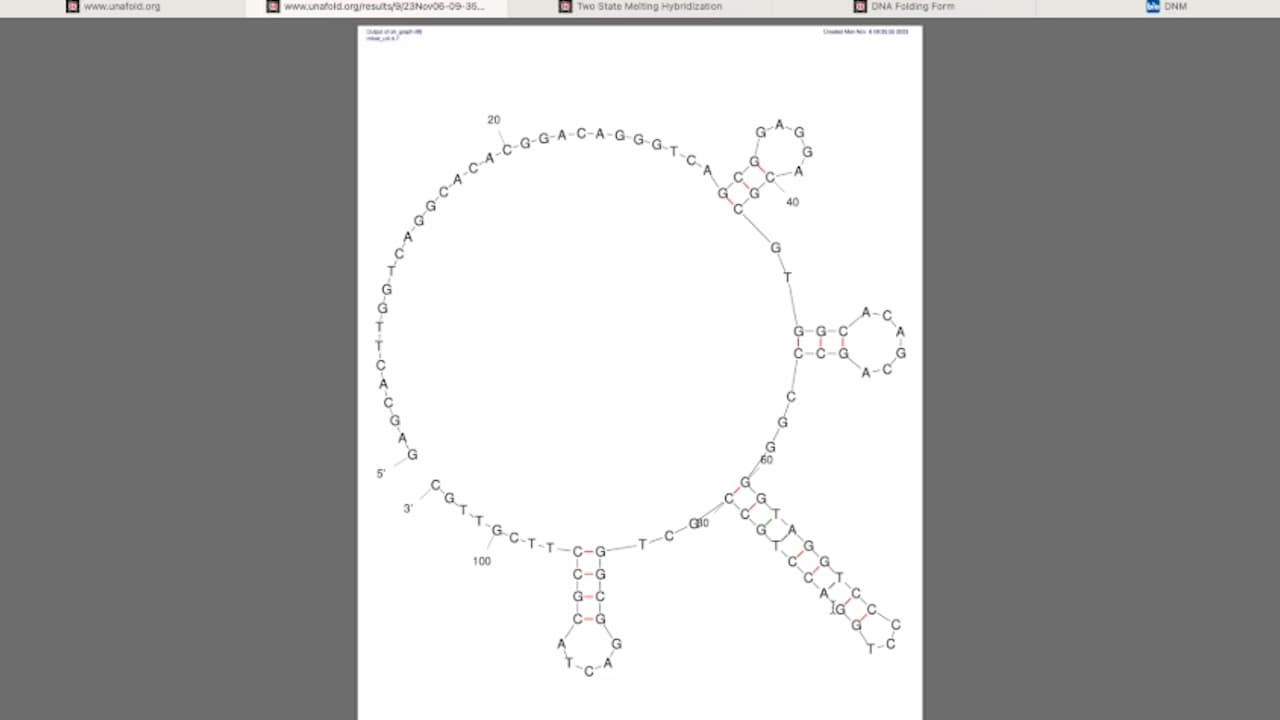
DNA has a double-helix structure. The...
DNA Structure
DNA has a double-helix structure. The...
 01:11
01:11Restriction Enzymes
Restriction enzymes are bacterial enzymes used to cut DNA in a sequence-specific manner. To cleave DNA, they bind to specific palindromic sequences called restriction sites. Such palindromic DNA sequences or inverted repeats are commonly found in regions of functional significance, such as the origin of replication, gene operator sites, and regions containing transcription termination signals.
The host bacteria protect their own genomic DNA from these enzymes by methylating these sites. Some...
The host bacteria protect their own genomic DNA from these enzymes by methylating these sites. Some...
 02:54
02:54Evolutionary Relationships through Genome Comparisons
Genome comparison is one of the excellent ways to interpret the evolutionary relationships between organisms. The basic principle of genome comparison is that if two species share a common feature, it is likely encoded by the DNA sequence conserved between both species. The advent of genome sequencing technologies in the late 20th century enabled scientists to understand the concept of conservation of domains between species and helped them to deduce evolutionary relationships across diverse...
 01:20
01:20Nucleic Acids and Nucleotides
Nucleic acids are the most important macromolecules for the continuity of life. They carry the cell's genetic blueprint and have instructions for its functioning. The two main types of nucleic acids are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA).
Deoxyribonucleic Acid (DNA)
DNA is the genetic material in all living organisms, ranging from single-celled bacteria to multicellular mammals. It is in the nucleus of eukaryotes and the organelles such as chloroplasts and mitochondria. In...
Deoxyribonucleic Acid (DNA)
DNA is the genetic material in all living organisms, ranging from single-celled bacteria to multicellular mammals. It is in the nucleus of eukaryotes and the organelles such as chloroplasts and mitochondria. In...
 01:57
01:57Sanger Sequencing
DNA sequencing is a fundamental technique that is routinely used in the biological sciences. This method can be applied to a range of questions at different scales - from the sequencing of a cloned DNA fragment or the study of a mutation in a gene up to whole-genome sequencing. However, despite the widespread use of sequencing today, it was not until 1977 that Fredrick Sanger and his collaborators developed the chain-termination method to decode DNA sequences. It relies on the separation of a...
 01:29
01:29Modern Molecular Taxonomy
Advancements in molecular biology have revolutionized the identification and characterization of bacteria, with multiple methods leveraging DNA sequencing for enhanced precision. As sequencing technologies improve and costs decline, these approaches are increasingly used in clinical, environmental, and evolutionary studies.Multilocus Sequence Typing (MLST) examines several housekeeping genes, essential chromosomal genes encoding cellular functions, to distinguish strains. Approximately...
You might also read
Related Articles
Articles linked to this work by shared authors, journal, and citation graph.
Sort by
Same author
A method for in silico exploration of potential glioblastoma multiforme attractors using single-cell RNA sequencing.
Scientific reports·2024
Same author
Structural Characterization of Heat Shock Protein 90β and Molecular Interactions with Geldanamycin and Ritonavir: A Computational Study.
International journal of molecular sciences·2024